
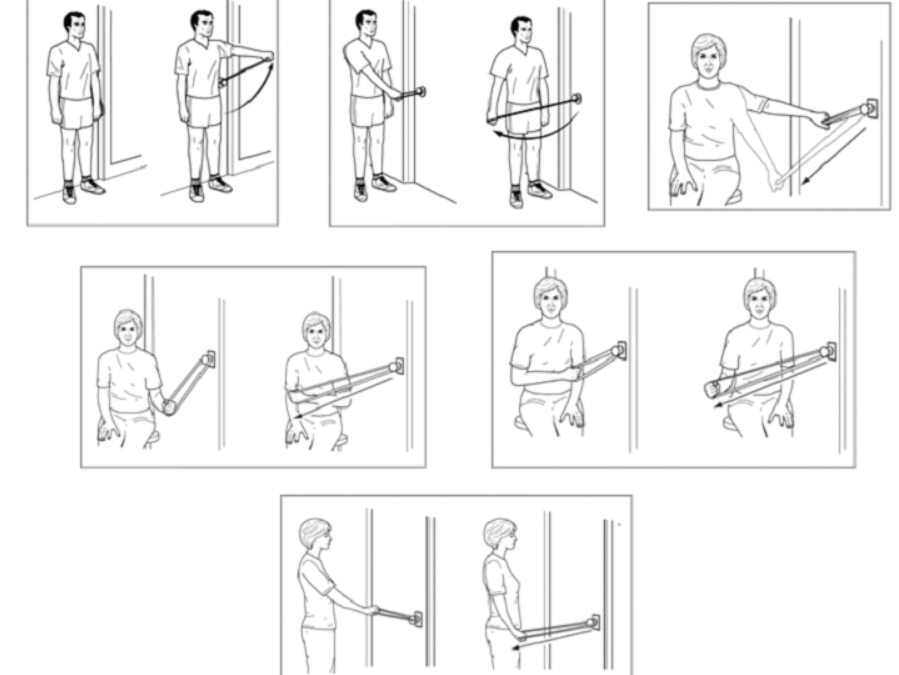
Ejercicios de Rehabilitación Física Posterior a Artroplastía (prótesis) de Rodilla
Ejercicios de Rehabilitación Física Posterior a Artroplastía (prótesis) de RodillaLos objetivos principales son: Ejercicios Temprano Posoperativos Ejercicios de Rehabilitación Física Posterior a Artroplastía (prótesis) de Rodilla El remplazo de rodilla con prótesis es...Asociación de lesiones del manguito rotador con dolor ycalidad del sueño. Una revisión sistemática
Esteban Castro Contreras M.D. Am J Med Surg February 2024; 15 (1). 45-51 IntroducciónObjetivoMetodologíaResultadosConclusionesDolor nocturno en pacientes con lesión del manguito rotador y dolor intensidadCalidad del sueño en pacientes con lesión del manguito...
Sirven las Inyecciones de Ácido Hialurónico para desgaste de rodilla
Uso de ácido hialurónicoÁcido Hialurónico para desgaste de rodilla Uso de ácido hialurónico El uso de ácido hialurónico ha mostrado ser efectivo en pacientes con osteoartrosis de rodilla hasta por 12 meses o más, se relaciona el beneficio del tratamiento cuanto...
Sirven las Células madre y Plasma Rico en Plaquetas para desgaste de rodilla
Plasma Rico en Plaquetas (PRP)Células madre y Plasma Rico Plasma Rico en Plaquetas (PRP) Este artículo trata sobre el uso de plasma rico en plaquetas (PRP) en aplicaciones ortopédicas y proporciona recomendaciones basadas en evidencia para su tratamiento. El PRP se...
¿Es útil el ejercicio en el desgaste de rodilla?
Osteoartritis de rodilla (KOA)ResultadosEjercició Aeróbico: Ejercicios de Fuerza: Ejercicios neuromusculares: Ejercicios de Balance. Ejercicios acuáticos: Osteoartritis de rodilla (KOA) La osteoartritis de rodilla (KOA) es una enfermedad común que causa dolor y...
¿Qué es la tenosinovitis de Quervain?
tenosinovitis de QuervainTratamiento tenosinovitis de Quervain La tenosinovitis de Quervain es una inflamación del primer compartimiento de la muñeca, afectando los tendones que controlan parte de los movimientos del pulgar. El origen de esta inflamación es...
¿Qué es la artroscopia de rodilla?
¿Qué es la artroscopia de rodilla? ¿Cómo se realiza una artroscopia de rodilla? ¿Qué es la artroscopia de rodilla?¿Cómo se realiza una artroscopia de rodilla? ¿Qué es la artroscopia de rodilla? La artroscopia de rodilla es un procedimiento sumamente habitual en la...
Lesiones de tendones y músculos
Estas lesiones pueden afectar a personas de todas las edades y niveles de actividad física, desde deportistas de alto rendimiento hasta aquellos que simplemente disfrutan de un estilo de vida activo. Las lesiones de tendones y músculos pueden deberse a una variedad de...
Que es el desgaste de rodilla o gonartrosis
¡Hola a todos! En este artículo les hablaré sobre una enfermedad que afecta a muchas personas en todo el mundo, especialmente a aquellas que ya han cumplido cierta edad: la gonartrosis. ¿Qué es gonartrosis?Cirugía de rodilla por gonartrosisCausas de...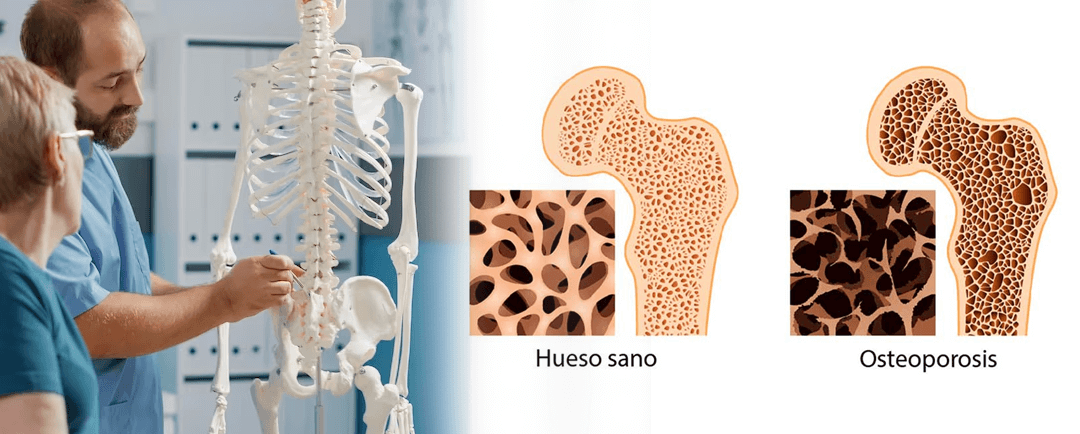
Consecuencias de la osteoporosis
¿Conocías las consecuencias de la osteoporosis? Aquí te mencionamos algunas consecuencias de la osteoporosis:¡No dudes en agendar una cita! ¿Conocías las consecuencias de la osteoporosis? La osteoporosis es una enfermedad silenciosa que debilita los huesos,...
Todo lo que el paciente debe de saber sobre la condromalacia
¿Has sido diagnosticado con Condromalacia, tienes dudas de que es y cómo se trata? Estas son algunas de las más frecuentes preguntas e inquietudes que tienen los pacientes: ¿Qué es la Condromalacia?¿Qué origina la Condromalacia?¿Que factores de riesgo pueden originar...
Lesiones deportivas más comunes
Lesiones deportivas más comunes Algunas de las lesiones deportivas más comunes son:Tratamientos Prevenir las lesiones deportivas Lesiones deportivas más comunes Hoy hablaremos sobre las lesiones deportivas. Estas son muy comunes en atletas y personas que realizan...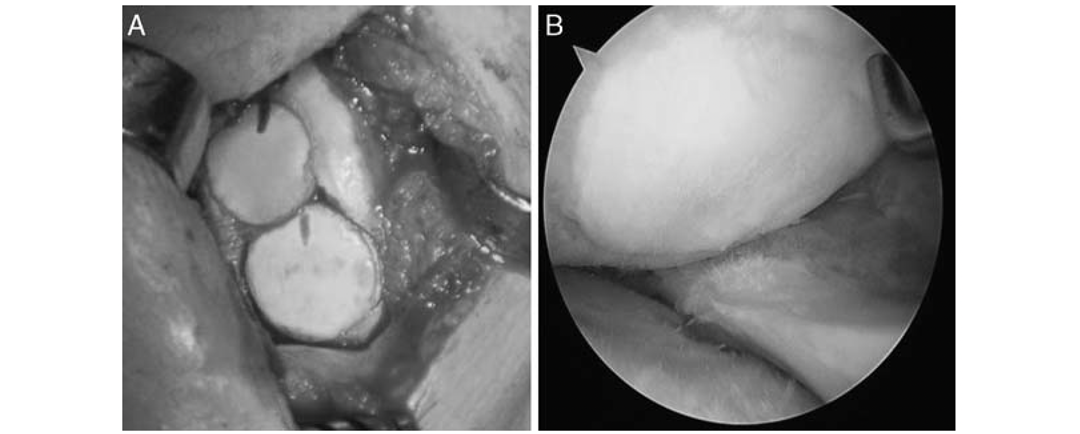
Restauración de lesiones complejas de cartílago
Restauración de lesiones complejas de cartílagoConclusiones Restauración de lesiones complejas de cartílago El artículo se enfoca en la lesión y degeneración del cartílago articular en atletas, lo cual puede llevar a una degeneración crónica de las articulaciones y...
¿Por qué truenan-crujen las articulaciones?
¿Por qué truenan-crujen las articulaciones?Conclusiones¿Te provoca dolor? ¿Por qué truenan-crujen las articulaciones? Si eres de los que presentan “crack – crack- crack” en la espalda o en las manos de forma voluntaria o involuntaria y te has vuelto una persona...
Movilización continua de rodilla
Movilización continua de rodillaConclusiones Movilización continua de rodilla El artículo presenta cinco estudios diferentes relacionados con la terapia de movimiento pasivo continuo (CPM) en pacientes que han recibido cirugía de reemplazo de rodilla o reconstrucción...
Injerto de cadaver vs autologo
Injerto de cadaver vs autologoResumen Injerto de cadaver vs autologo El artículo se enfoca en comparar los resultados de la reconstrucción del ligamento cruzado anterior (ACL, por sus siglas en inglés) utilizando dos tipos de injertos: tendón de la corva autólogo y...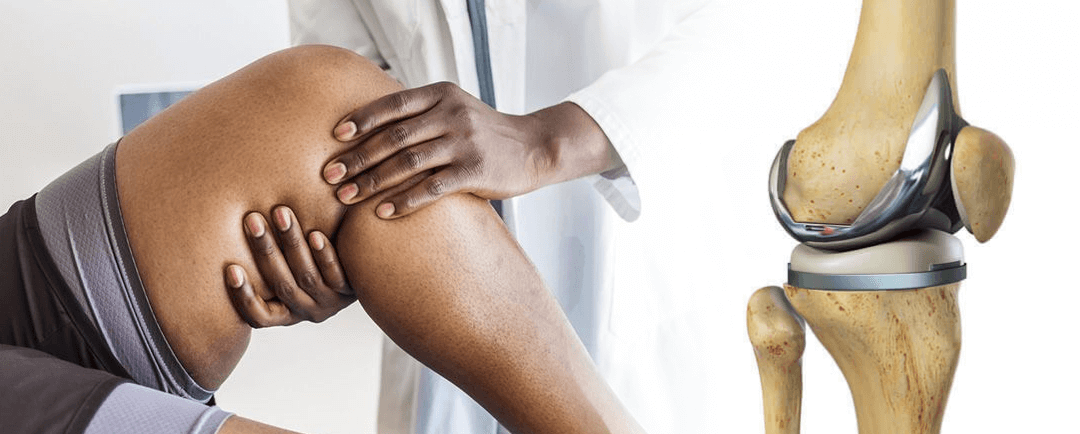
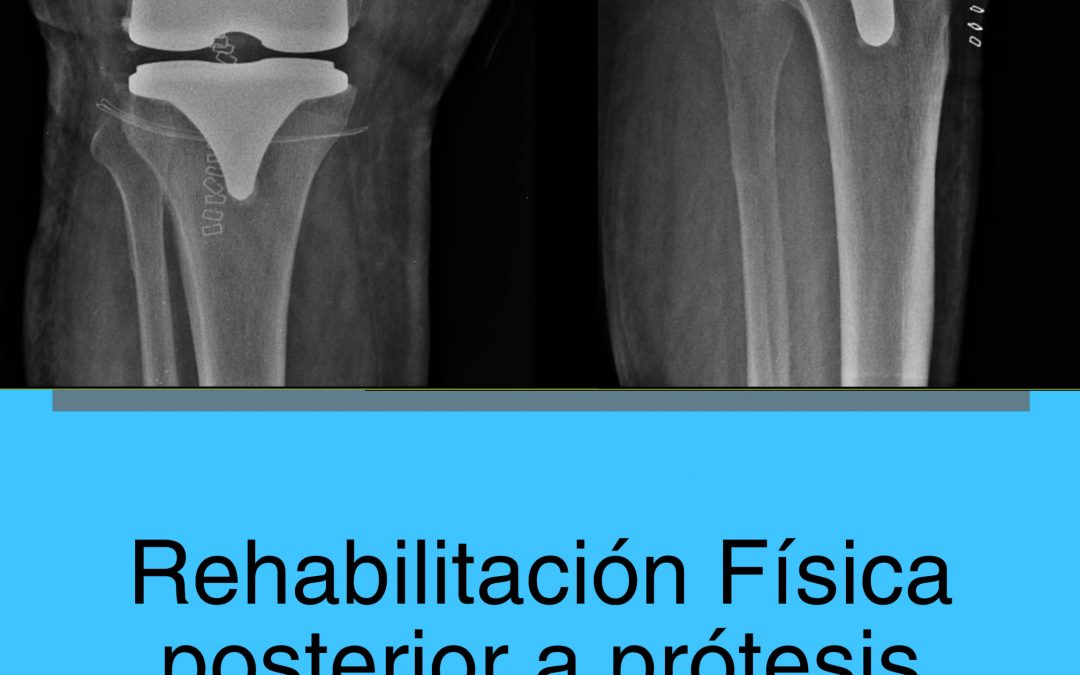
Cuanto dura una protesis de rodilla
Cuanto dura una protesis de rodillaConclusiones Cuanto dura una protesis de rodilla Un nuevo estudio publicado en la revista médica The Lancet ha encontrado que las prótesis de rodilla duran al menos 25 años en la mayoría de los pacientes. El estudio analizó datos de...
Todo lo que el paciente debe de saber de las lesiones deportivas
Lesiones deportivasPuntos importantes sobre las lesiones deportivas¿Quién puede sufrir una lesión deportiva?¿Que son las lesiones Deportivas?¿Cuáles son los factores de riesgo para una lesión deportiva?¿Qué tipos de lesiones Deportivas existen?Síntomas de una lesión...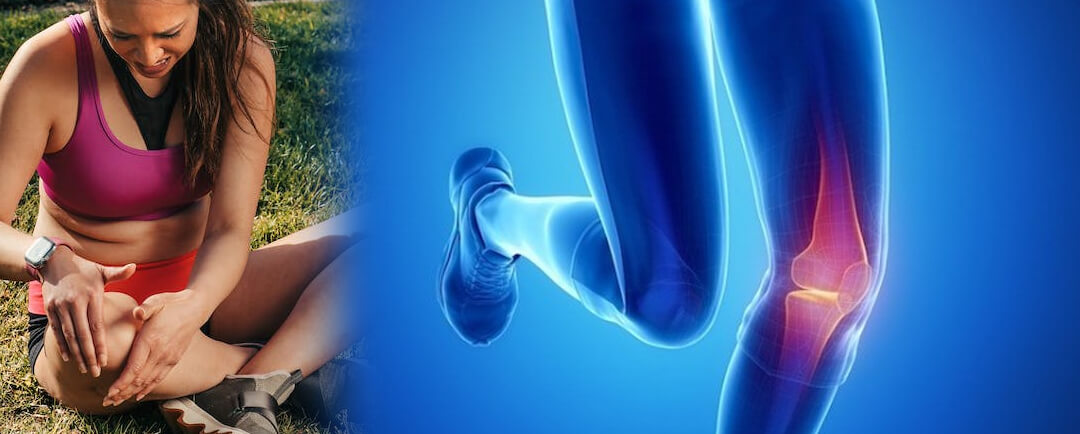
Cirugía de ligamento cruzado anterior
Table of Contentscirugía de ligamento cruzado anterior¿Cómo se lesiona el ligamento cruzado anterior de la rodilla?¿Qué síntomas produce una lesión de Ligamento cruzado anterior de rodilla?¿Cómo se diagnostica una lesión de ligamento y quien debería de hacer ese...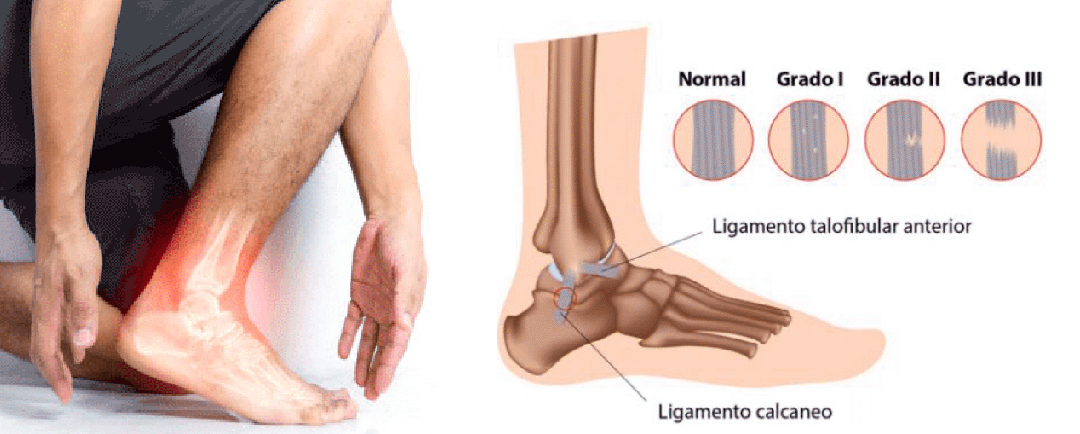
Esguince de tobillo
Table of ContentsSíntomas de un esguince de tobillo:Tipos de esguinces de tobilloEl tratamiento de un esguince de tobillo consiste en: Por último, para prevenir los esguinces de tobillo: ¿Alguna vez te has hecho un esguince? Hablemos un poco sobre ellos. Un esguince...
Conoce las causas del dolor de huesos y como prevenirlo
El dolor de huesos es una molestia que puede afectar a cualquier persona, en cualquier edad y por diversas razones. Si estás experimentando dolor de huesos, es importante conocer las causas más comunes para poder tomar medidas para prevenirlo o tratarlo adecuadamente....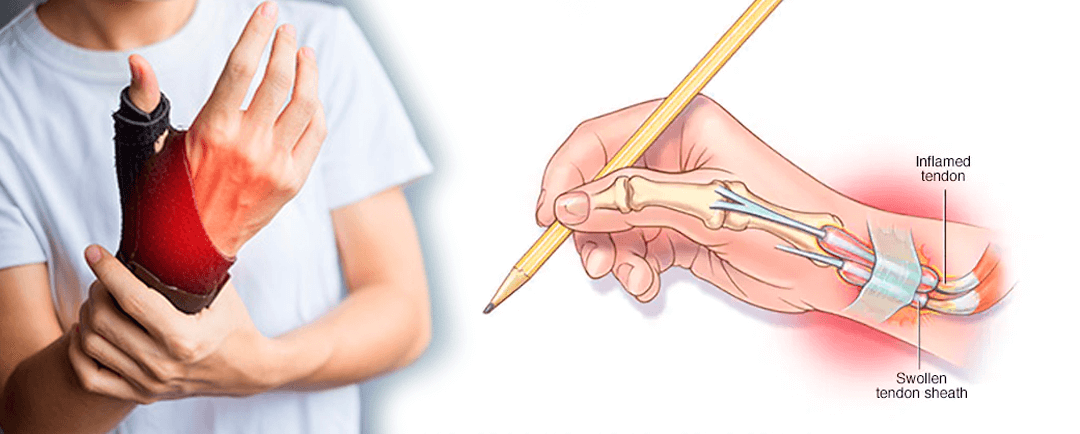
Tenosinovitis de De Quervain
Tenosinovitis de de QuervainLos síntomas de la tenosinovitis de De Quervain comprenden los siguientes:Cuándo consultar al médicoCausasFactores de riesgoComplicaciones Tenosinovitis de de Quervain La tenosinovitis de De Quervain es una inflamación de los tendones de la...
Sobrevida de rodillas protésicas
Sobrevida de rodillas protésicasLos resultados de este estudio muestran que: Sobrevida de rodillas protésicas Una de las dudas mas frecuentes en el paciente quien requiere un reemplazo articular de rodilla o de otras regiones de la economía articular es ¨¿Cuánto...
Manejo agudo de esguinces de tobillo
Manejo agudo de esguinces de tobilloSe señalan 3 fases de re integración Manejo agudo de esguinces de tobillo 80% de los individuos a lo largo de su vida sufriran un esguince de tobillo. Los deportes a puerta cerrada (Voleibol, Basquetbol, danza, por ejemplo) son los...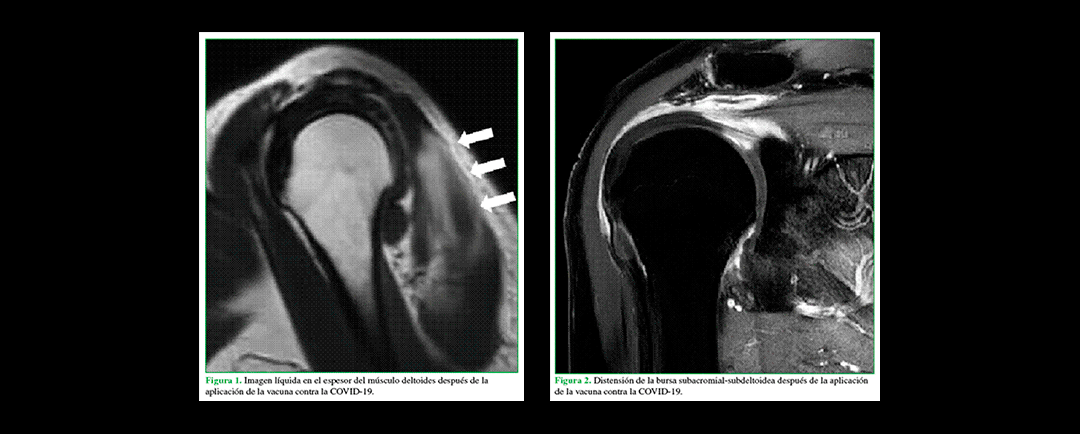
Lesiones de hombro atribuibles a la aplicación de la vacuna contra la COVID-19
Lesiones de hombro atribuibles a la aplicación de la vacuna contra la COVID-19Características: Lesiones de hombro atribuibles a la aplicación de la vacuna contra la COVID-19 Las lesiones de hombro atribuibles a la vacunación son aquellos efectos adversos secundarios a...
Los efectos del COVID-19 sobre la morbilidad y mortalidad perioperatoria en pacientes con fracturas de cadera
Los efectos del COVID-19 sobre la morbilidad y mortalidad perioperatoria en pacientes con fracturas de cadera Table of ContentsOBJETIVOMATERIAL Y MÉTODOSRESULTADOSCONCLUSIÓN OBJETIVO Durante la pandemia de COVID-19, muchos pacientes continúan requiriendo cirugía...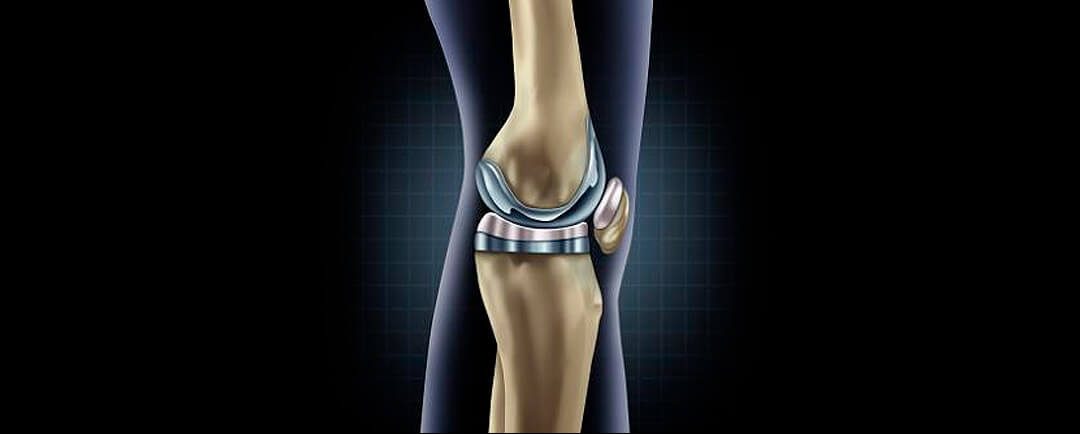
Las inyecciones de corticosteroides intraarticulares aumentan el riesgo de requerir una artroplastia de rodilla
Las inyecciones de corticosteroides intraarticulares aumentan el riesgo de requerir una artroplastia de rodillaOBJETIVOMATERIAL Y MÉTODOSRESULTADOSCONCLUSIÓN Las inyecciones de corticosteroides intraarticulares aumentan el riesgo de requerir una artroplastia de...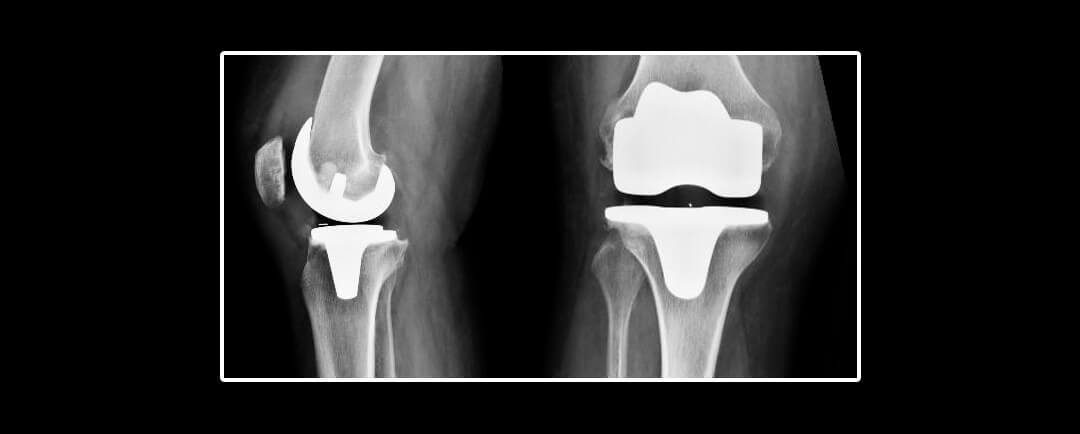
Complicaciones tromboembólicas y hemorrágicas después de una artroplastia total de rodilla primaria: Un estudio de cohorte nacional danés
Complicaciones tromboembólicas y hemorrágicas después de una artroplastia total de rodilla primaria: Un estudio de cohorte nacional danésOBJETIVOMATERIAL Y MÉTODOSartroplastia total de rodillaRESULTADOSCONCLUSIÓN Complicaciones tromboembólicas y hemorrágicas después...
Afección muscular y articular en pacientes post COVID-19
Afección muscular y articular en pacientes post COVID-19 INTRODUCCIÓNOBJETIVOMATERIAL Y MÉTODORESULTADOS DISCUSIÓN Bibliografía: INTRODUCCIÓN El SARS-Cov-2 es una enfermedad pandémica emergente que causa el síndrome respiratorio agudo severo. Aunque la mayoría de los...


María Velasco
19:57 16 Mar 23
Hola buenas tardes, quiero agradecer al Dr. Esteban Castro su gran dedicación , en lo personal mi cirugía fue un éxito me siento muy bien, es un excelente Doctor q nos brinda confianza y atencion

JESUS MONTES LEON
22:00 15 Mar 23
La atención muy bien...El detalle que no me pareció es que si ya tienes agendada una cita me la hayan cambiado primero de día...Y después la hora...Quiero aclarar que ya había hecho la cita aproximadamente hace un mes...

carlos cisneros
00:55 07 Mar 23
Buena atención aún desde telefónica como presencial del personal de recepción. El doctor, muy involucrado en su atención hacia un servidor, así como oportuna toma de desiciones después de algunas otras alternativas y al ver que no hubo mejoría, tomó la desicion de realizar una artroscopia de hombro izquierdo . Toda mi fe y confianza en él pues sería la segunda cirugia que me realice, la otra fue en el hombro derecho hace año y medio, de lo que quedé muy bien. Enhorabuena.

Silvia Sanchez
23:11 22 Feb 23
Dr. Esteban Castro, para mi, es el mejor traumatólogo, me practicó una cirugía de hombro, la cuál considero que la realizó en forma excelente, no presente ninguna molestia, y estoy recuperándome muy bien, mil gracias Dr. y estoy muy agradecida por sus atenciones y por ayudarme a recuperar mi salud. Dios lo bendiga.

Kari Morones Armenta
18:50 21 Feb 23
Excelente médico, a mí me operó las dos rodillas y hasta ahorita genial. Muy recomendado y la atención brindada por él y su equipo.

Hector Martinez Jr.
19:00 07 Feb 23
Fui por una consulta y me base en las reseñas. Al comienzo el doctor me dijo q basado a el problema q le explique q era una cosa. Después de 17,000 pesos de estudios me dijo q el problema q tengo en mis hombros era una deterioración muscular genética de la cintura y q tenia q irme a ver un neurólogo para hacerme otros estudios.Fui por una segunda opinión en donde revisaron los estudios q el Dr. Esteban me mando hacer. Y basado a esos estudios me dijeron q tenia una hernia en un disco empujando la medula en el cuello y opérculo torácico como el problema en mis hombros. Para estar bien seguros de la cirugía me pidieron otros estudios ya q a simple vista el problema q tengo es obvio y los estudios q me mandaron hacer fueron generales no específicos al problema o el lugar problemático en mi cuello y hombros. $15,000 pesos mas.Me fui inconforme con el diagnostico de un problema de la cintura ya q el problema q explique era del cuello y los hombros. No lo recomendaría.

Roberto Espinoza
04:31 21 Jan 23
Excelente médico, trato muy amable y capacitado mi cirugía a pesar de ser muy complicada resultó un éxito.

Lau
19:25 13 Jan 23
Muy buena atención por parte del doctor, muy profesional. Resolvió todas mis dudas y me indicó el tratamiento necesario.Lo único que llegó algo tarde pero valió la pena totalmente.

Gema Araceli Angel Rodriguez
02:44 07 Jan 23
Excelente Médico trato humano y sobre todo seguimiento puntual a sus pacientes felicidades.

Martha Alicia Juárez
20:50 03 Jan 23
Excelente doctor, muy capacitado. Su trato suele ser amable. Explícito en sus comentarios y resuelve dudas.

Lupita Franco
20:34 02 Dec 22
Excelente doctor y gran persona y si me explico muy bien el problema que yo tengo

Alba Hernández
01:07 03 Nov 22
Recomiendo mucho al doctor Esteban, hace unos días me realizo una artroscopia de rodilla, puedo decir que ha sido el ángel que dio con mi diagnóstico después de casi dos años viviendo con dolor, vueltas con varios traumatólogos y estudios y una vida con muchas limitaciones. El doctor desde el primer día en consulta me transmitió esa confianza y no me operó hasta tener la seguridad de que pasaba con mis rodillas, siempre fue muy certero con toda la sintomatología que presentaba. Es una persona muy profesional y muy humano, sobre todo el trato con los pacientes, muy amable siempre en todas las consultas hasta el día de mi cirugía. Estoy en proceso de recuperación pero después de la artroscopia me he sentido mucho mejor, el dolor por mi padecimiento desapareció, solo es el malestar de la intervención, y puedo decir que definitivamente estuve en la mejores manos. Y doy muchas gracias por caer en manos de un excelente doctor.

fernando godinez
22:08 26 Oct 22
Excelente Traumatologo el Dr. Esteban Castro, Con mucha Ética Profesional. La verdad es una eminencia, a mi me opero de hombro y rodilla y estoy completamente satisfecho con su trabajo, quede muy bien de mi hombro y de mi rodilla, Dios te bendiga Dr. Esteban Castro.

Lyzeth Guzman
23:38 23 Oct 22
Increíble mi experiencia con el doctor, paciente a responder todas mis dudas súper bien explicado, todo el personal fue súper amable , súper recomendado


Karen Meza Velazquez
01:39 15 Oct 22
Excelente doctor, te explica a detalle tu caso y tratamiento, muy paciente y lindo. Me hicieron sentir segura, no sólo el, si no todo su personal...

silvia garcia
15:57 09 Oct 22
Excelente atención, siempre al pendiente de mi padecimiento y muy acertado. Gracias Doctor

Miguel Angel Mendoza Navarrete
00:30 01 Oct 22
Super agradecido con el Doctor muy buena atención, sus recomendaciones muy bien explicadas y se nota que es una persona con buenos valores.

Lorena Quezada Frias
17:30 21 Sep 22
Excelente trato de todo el personal desde que llegas.Explica súper bien el tratamiento, pasos a seguir, opciones de tratamientos y aclara todas tus dudas.Instalaciones en súper buen estado y buena ubicación.

JESUS ANTONIO ALEJANDRE TORRES
05:29 05 Aug 22
100% satisfecho y con toda la confianza depositada en el doctor Esteban Castro, a casi un mes de mi cirugía de columna me siento motivado para dar el siguiente paso a la rehabilitación.

ananias tobiel
02:45 05 Aug 22
Soy un paciente del doctor..Esteban..pase por una cirujia..y ahora ando en proceso de rehabilitación...les comparto mi vivencia durante el proceso..es una excelente persona tanto como dentro y fuera del hospital...y de sus labores excelente trabajo lo que el realiza...muy seguro de lo que hace..recomiendo su trabajo.. y te atienden de una manera muy buena y amable...

Julia Jacqueline Guillen
21:28 04 Aug 22
Es un especialista altamente preparando .... Su trabajo habla por si sólo.Confiable , Profesional y Responsable .Gracias Doctor Castro 🤝

Alan O Olmedo G
02:04 04 Aug 22
El Dr. Esteban Castro es un muy buen cirujano, hace tiempo operó a un familiar mio y le fue de maravilla, nos resolvió nuestras dudas paso a paso y nos brindo una excelente atención al igual que todo su equipo medico. Totalmente recomendable.

Dra. Daniela Bañuelos
22:20 27 Jul 22
Excelente atención médica; excelentes resultados en mi cirugía. Además de buen médico buen ser humano; eso vale mucho más! Gracias Esteban!!!!!



Eduridan23
03:35 29 Aug 21
Estoy muy agradecido con el doctor por su profesionalismo y tiempoMuy recomendado y atención al 100

ANA PAOLA ORNELAS LEAL
18:33 20 Aug 21
Un excelente médico, me encantó su manera de tratar con el paciente.

Enrique Contreras
04:33 01 Jul 21
Bueno, eficaz y rápido, trato bueno y muy amable. Te hace sentir seguro.

Ana Luisa Vazquez Ortiz
21:32 24 Apr 21
Fui por una lesión en la espalda y el doctor super profesional y atento, muy acertado en su diagnósticoRecomendado 100%


Juan francisco Gonzalez Rodriguez
04:27 26 Feb 21
Muy buen doctor, gracias a su consulta mi padecimiento se corrigio. Estoy muy satisfecho.

Valentin Organes
19:35 17 Feb 21
Muy atento y claro. Me explico con paciencia el por qué de todo mi malestar. Recomendado.